Software Crystal Impact Endeavour
SKU: Endeavour




SKU: Endeavour
Interface Inovadora
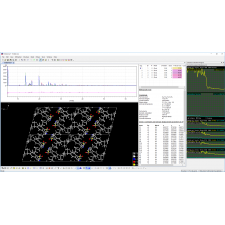
A interface inovadora e intuitiva torna o processo de solução de estruturas cristalinas quase rotineiro, especialmente para compostos inorgânicos, mas também para muitos compostos orgânicos.
Facilidade de Uso
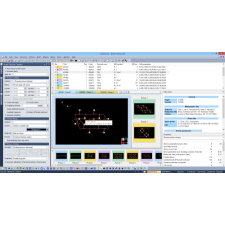
Mesmo usuários inexperientes podem preparar e executar cálculos de solução de estrutura em poucos passos. O assistente integrado guia o usuário na inserção dos dados necessários (parâmetros da célula unitária, composição química e dados de difração), permitindo que o Endeavour faça o restante.
Método de Solução
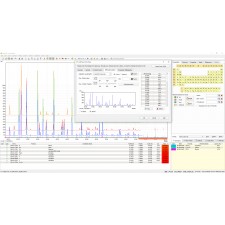
A solução de estrutura é realizada por meio de uma variante especial do método "direto-espaço", que combina a otimização global da diferença entre dados de difração calculados e observados, além da energia potencial do sistema. Este método é menos sensível a dados de difração de baixa qualidade em comparação com métodos diretos.
Atualizações da Versão 1.8
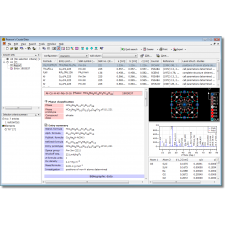
A versão mais recente, 1.8, traz uma interface de usuário redesenhada, além de várias melhorias e correções de bugs.