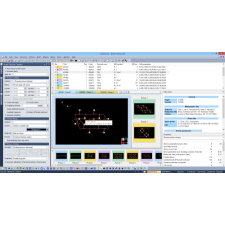
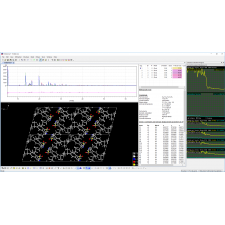
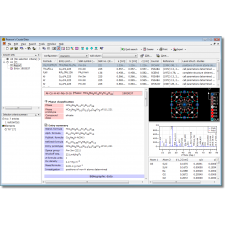
Match! é um software intuitivo para identificação de fases a partir de dados de difração de pó.
Ele compara o padrão de difração da sua amostra com um banco de dados contendo padrões de referência, permitindo a identificação das fases presentes. Informações adicionais sobre a amostra, como fases conhecidas, elementos ou densidade, podem ser facilmente aplicadas.
Análise Qualitativa e Quantitativa
Além da análise qualitativa, uma análise quantitativa pode ser realizada utilizando o refinamento Rietveld. O Match! permite configurar e executar refinamentos Rietveld com facilidade, utilizando o renomado programa FullProf (de J. Rodriguez-Carvajal) em segundo plano. O software proporciona uma introdução gradual ao refinamento Rietveld, desde a operação totalmente automática até o modo "Expert". O Match! é compatível com Windows, macOS e Linux.
Banco de Dados Referencial
Você pode usar o banco de dados COD incluído gratuitamente, qualquer produto ICDD PDF e/ou criar um banco de dados de usuário com base em seus próprios padrões de difração. Os padrões do banco de dados do usuário podem ser editados manualmente, importados de arquivos de picos, calculados a partir de dados de estrutura cristalina (por exemplo, arquivos CIF) ou importados do banco de dados de um colega.
Principais Funcionalidades
- Refinamento Rietveld (usando FullProf): Transfira seus dados com apenas dois cliques e realize um refinamento Rietveld.
- Indexação: Utilize os programas de indexação mais renomados, como Treor90 e Dicvol06, para determinar a célula unitária.
- Solução de Estruturas: Resolva estruturas a partir de dados de difração de pó usando o pacote “Endeavour”.
- Compatibilidade Multi-Plataforma: O Match! roda em Windows, macOS e Linux, permitindo o uso de arquivos em qualquer sistema operacional.
- Comparação de Padrões de Difração: Importe e exiba padrões experimentais adicionais sobrepostos para comparações.
- Definição de Background e Zoom Melhorado: Ajuste facilmente o fundo e faça zoom nas áreas de interesse de forma intuitiva.
- Processamento em Lote e Automático: Adapte a operação ao seu nível de habilidade, desde controle total até automação completa.
- Estimativa do Tamanho de Cristalitos: Calcule os valores do tamanho de cristalitos com base nos dados do pico utilizando a fórmula de Scherrer.
Requisitos do Sistema (mínimos)
- Windows: PC com Windows XP, Vista, 7, 8 ou 10; 2.5 GB de RAM; 500 MB de espaço livre; resolução gráfica mínima de 1024 x 768 pixels.
- macOS: Mac com processador Intel e macOS 10.12 ou superior; 2.5 GB de RAM; 500 MB de espaço livre; resolução gráfica mínima de 1024 x 768 pixels.
- Linux: PC com Linux (Intel 64-bit); 2.5 GB de RAM; 500 MB de espaço livre; resolução gráfica mínima de 1024 x 768 pixels.